Abstract
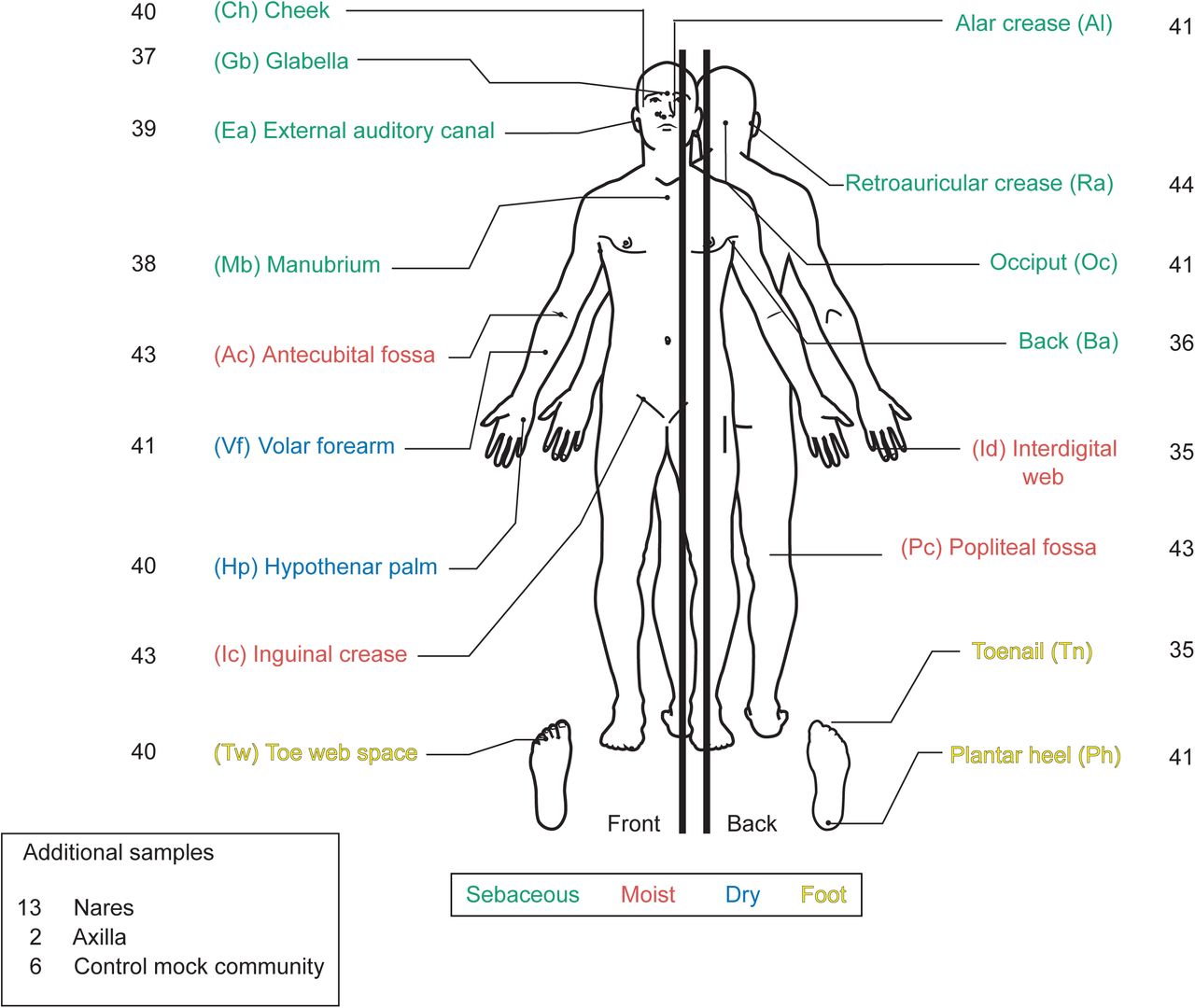
The healthy human skin microbiome is shaped by skin site physiology, individual-specific factors, and is largely stable over time despite significant environmental perturbation. Studies identifying these characteristics used shotgun metagenomic sequencing for high resolution reconstruction of the bacteria, fungi, and viruses in the community. However, these conclusions were drawn from a relatively small proportion of the total sequence reads analyzable by mapping to known reference genomes. ‘Reference-free’ approaches, based on de novo assembly of reads into genome fragments, are also limited in their ability to capture low abundance species, small genomes, and to discriminate between more similar genomes. To account for the large fraction of non-human unmapped reads on the skin—referred to as microbial ‘dark matter’—we used a hybrid de novo and reference-based approach to annotate a metagenomic dataset of 698 healthy human skin samples. This approach reduced the overall proportion of uncharacterized reads from 42% to 17%. With our refined characterization, we revisited assumptions about the skin microbiome, and demonstrated higher biodiversity and lower stability, particularly in dry and moist skin sites. To investigate hypotheses underlying stability, we examined growth dynamics and interspecies interactions in these communities. Surprisingly, even though most skin sites were relatively stable, many dominant skin microbes, including Cutibacterium acnes and staphylococci, were actively growing in the skin, with poor or no relationship between growth rate and relative abundance, suggesting that host selection or interspecies competition may be important factors maintaining community homeostasis. To investigate other mechanisms facilitating adaptation to a specific skin site, we identified Staphylococcus epidermidis genes that are likely involved in stress response and provide mechanisms essential for growth in oily sites. Finally, horizontal gene transfer—another mechanism of competition by which strains may swap antagonistic or virulent coding regions—was relatively limited in healthy skin, but suggested exchange of different metabolic and environmental tolerance pathways. Altogether, our findings underscore the value of a combined reference-based and de novo approach to provide significant new insights into microbial composition, physiology, and interspecies interactions to maintain community homeostasis in the healthy human skin microbiome.