Abstract
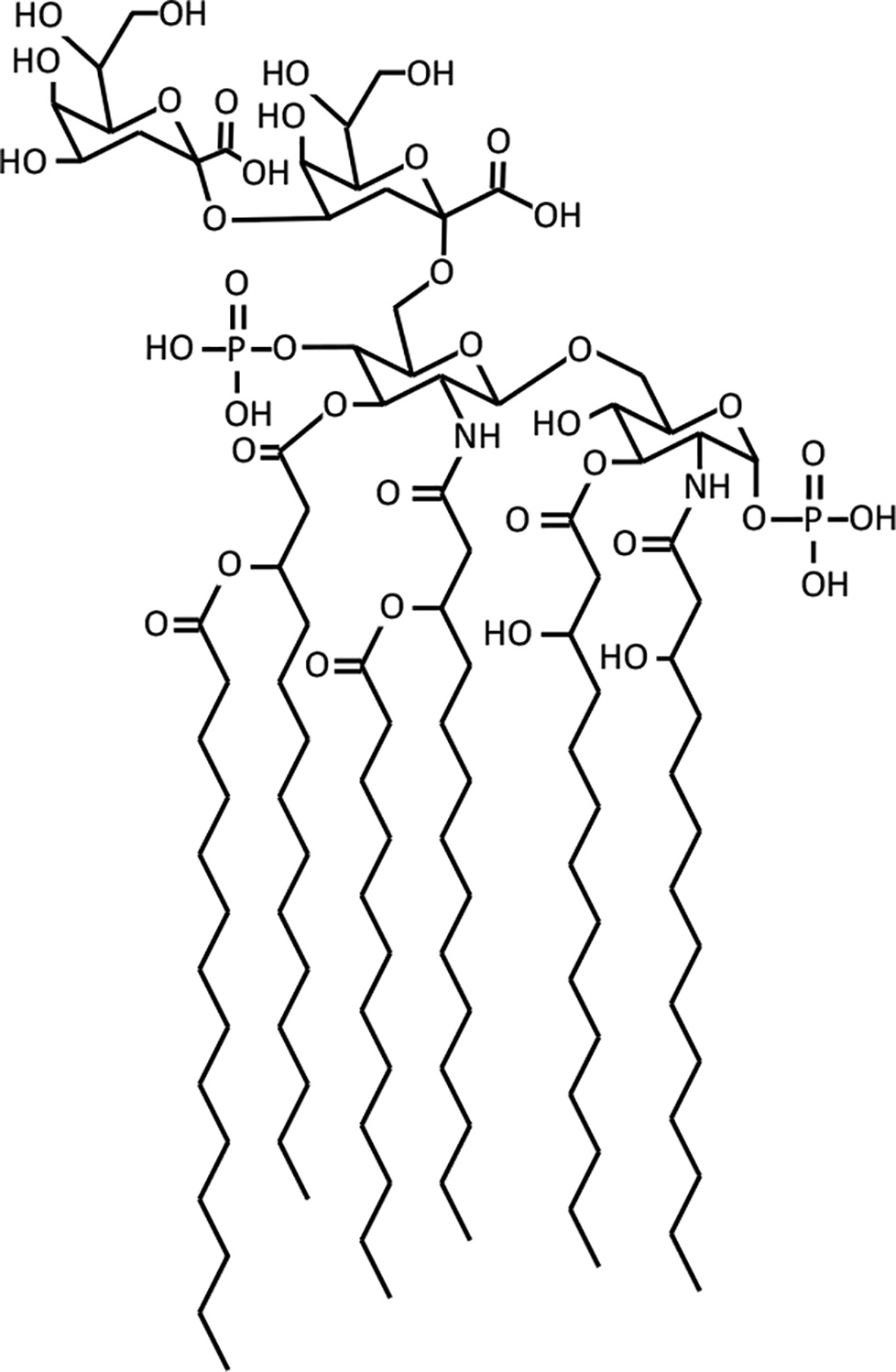
Lipid A is a highly conserved component of lipopolysaccharide (LPS), itself a major component of the outer membrane of Gram-negative bacteria. Lipid A is essential to cells and elicits a strong immune response from humans and other animals. We developed a quantitative model of the nine enzyme-catalyzed steps of Escherichia coli lipid A biosynthesis, drawing parameters from the experimental literature. This model accounts for biosynthesis regulation, which occurs through regulated degradation of the LpxC and WaaA (also called KdtA) enzymes. The LpxC degradation signal appears to arise from the lipid A disaccharide concentration, which we deduced from prior results, model results, and new LpxK overexpression results. The model agrees reasonably well with many experimental findings, including the lipid A production rate, the behaviors of mutants with defective LpxA enzymes, correlations between LpxC half-lives and cell generation times, and the effects of LpxK overexpression on LpxC concentrations. Its predictions also differ from some experimental results, which suggest modifications to the current understanding of the lipid A pathway, such as the possibility that LpxD can replace LpxA and that there may be metabolic channeling between LpxH and LpxB. The model shows that WaaA regulation may serve to regulate the lipid A production rate when the 3-deoxy-D-manno-oct-2-ulosonic acid (KDO) concentration is low and/or to control the number of KDO residues that get attached to lipid A. Computation of flux control coefficients showed that LpxC is the rate-limiting enzyme if pathway regulation is ignored, but that LpxK is the rate-limiting enzyme if pathway regulation is present, as it is in real cells. Control also shifts to other enzymes if the pathway substrate concentrations are not in excess. Based on these results, we suggest that LpxK may be a much better drug target than LpxC, which has been pursued most often